

前天,一份CDFA出台的征求意见稿在肿瘤临床领域引起了掀然大波,引得无数医药人振奋:如意见顺利实施,中国的药品临床制度将大大提升效率,加快临床新药审批速度;还有部分肿瘤患者能借此提前用上抗癌“救命药”。

到底是什么制度如此实现了这样的优化,首先还要从CFDA的职责说起:
CFDA全称为中国食品药品监督管理,负责药品、医疗器械、化妆品和消费环节食品安全的监督管理。但对于各位患友而言,其最重要的职责就是负责审批提交上市申请的临床新药,根据这些新药进行的一期、二期、三期临床试验数据决定是否允许该药物上市。也就是说:一个抗肿瘤新药在中国的上市与CFDA的审批效率密切相关。
谈起这两年来肿瘤临床新药的进步,不仅是临床医生,就连患者们也要竖起大拇指:短短几年间,先后有PD-1抑制剂和几种“重量级”的靶向药面市,这些临床新药为肿瘤的治疗带来了划时代的飞跃:它们取得了横跨多癌种的突破性进展,部分晚期患者因这些新药而长期生存,甚至临床治愈。
然而这些“救命”的新药却在中国陷入了两难:由于人种差异和一些原因,国外已经完成临床试验并成功上市的抗肿瘤新药来到中国依然需要进行漫长的审批与临床试验。这个时间通常需要三年、五年甚至更久。这个原因导致了中国肿瘤的治疗水平与世界的顶尖水平拉开了一定差距,中国患者也只能眼看着可以“救命”的新药处于审批的过程中而无法使用。
漫长的审批在近两年发生了转变。随着国家对医疗体系的逐步重视,CFDA正不断完善药物审批流程,推进一些重要药物的上市进程。
今年,两款肺癌最“重磅”的靶向药奥希替尼(AZD9291)与阿法替尼(2992)先后完成了上市,最受期待的“抗癌神药”PD-1抑制剂也先后获得审批,预计2018年上半年就可完成上市。
紧接着,CFDA又开挂了!
12月20日,CFDA发布征求意见稿,拟在药物临床审批制度中推行拓展性同情使用临床试验用药物管理办法。
简单来说,就是急需使用临床新药的患者可以通过向临床试验方(也就是药厂方)提交申请,经临床试验方通过后,即可在不符合入组标准的条件下参与临床试验,获得临床新药的治疗。
这一意见对于临床新药的上市审批与患者而言都有非常大的优势:由于大部分临床试验的入组条件都较为苛刻,不是所有能因新药受益的患者都有入组的条件。而意见一旦实施后,通过咚咚临床招募平台,部分不满足临床要求的患者也可通过本条款申请用药,大大提高了入组临床的可能性,及时用上新药,有时甚至意味着关键时刻的病情逆转!
咚咚临床招募计划
关于临床招募计划,已经不是第一次在公众微信号里与大家提到了。相信各位对这个可以免费用上“救命”抗癌药的福利都有一定程度的了解。不熟悉的癌友可以点击右边链接:抗癌神药免费“派送”丨临床试验,先到先得
事实上,临床实验并不适用于所有癌友,我们不推荐具有明确治疗方案的患者选择参与临床。但国内外几乎所有的专家,对于参与临床可能显著获益的患者及没有标准治疗方案的患者,都会积极推荐参与临床实验。而这恰与CFDA拟实施的拓展性同情使用临床药物的意见高度吻合。面临可能显著获益的临床药物,即使不符合条件的患者也可通过咚咚招募平台,积极争取用上临床新药的机会。
特别是在抗肿瘤新药不断更新的今天,很多临床实验几乎等同于用上最新药物的机会,成为了癌友们的打败病魔的制胜绝招。
需要进一步了解咚咚临床招募的患友们,可以联系咚咚临床科研助手进行咨询。
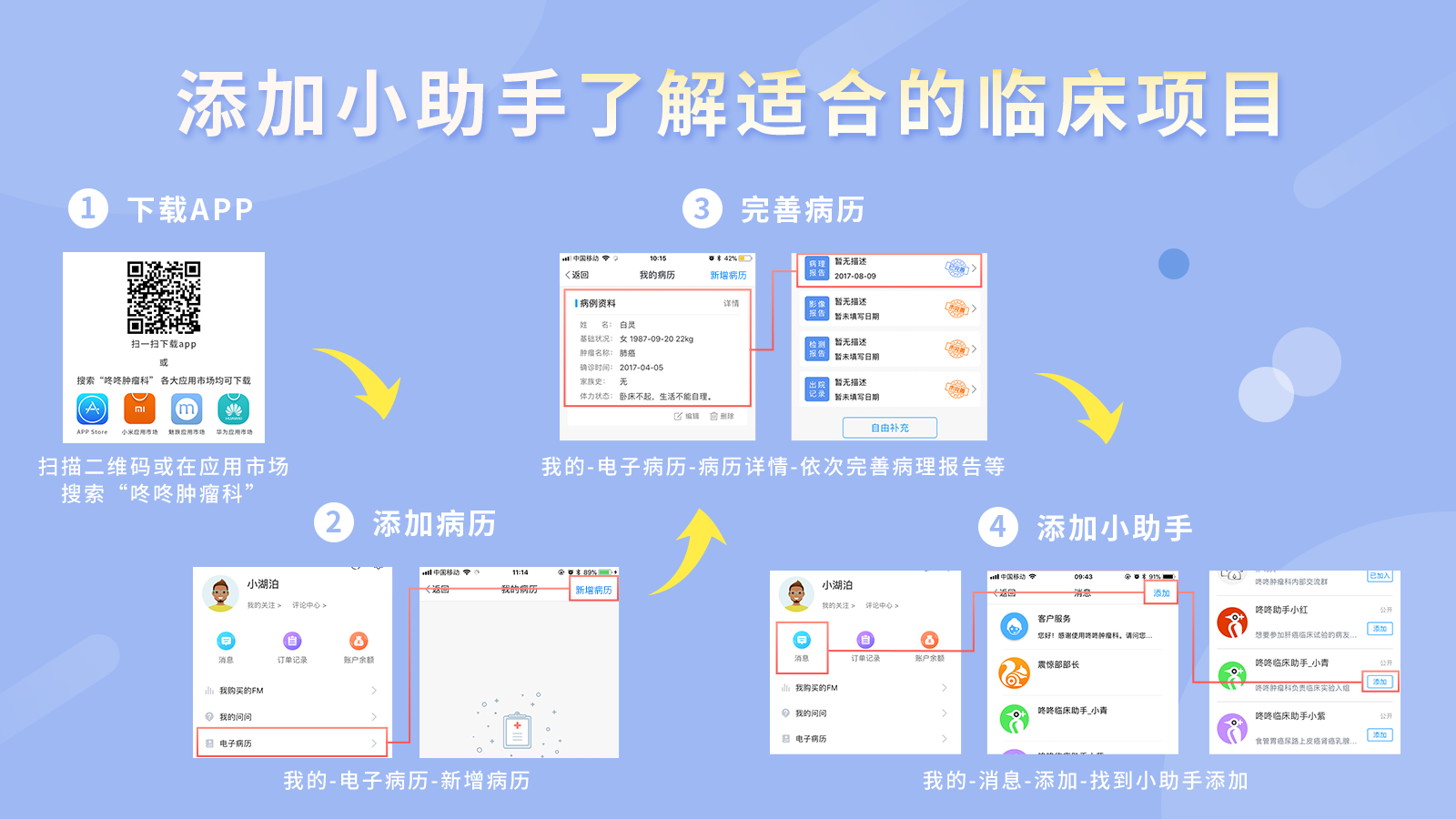
最后附上CFDA颁布的拓展性同情使用临床试验用药物管理办法(意见稿),可以点击链接:http://www.sfda.gov.cn/WS01/CL0778/219885.html,去CFDA官网下载word文档,如果有什么意见,可以于2018年1月15日前,将意见反馈至电子邮箱zhaochy@cde.org.cn。
本文仅供医学药学专业人士阅读



-1.jpg-pd13)
-1-scaled.jpg-pd13)
.jpg-pd13)













 X
X