
2021即将落幕,一年一度的年度盘点如约而至,在这一年里,乳腺癌领域都有那些不容错过的进展,一起跟随小编的步伐看一下吧~
关口前移,奥拉帕利进军早期乳腺癌,
将复发风险降低42%!
2021年12月1日 ,FDA 授予奥拉帕利一项补充新药申请 (sNDA) 优先审评资格,用于BRCA突变、高风险、HER2 阴性早期乳腺癌患者的辅助治疗,这些患者以前接受过新辅助治疗或辅助化疗。
该申请基于 OlympiA 3 期试验 (NCT02032823) 的结果,该结果在 2021 年 ASCO 年会上提出,并同时发表在新英格兰医学杂志上。
OlympiA 研究是一项多中心、随机、安慰剂对照试验,共招募了 1836 例携带种系BRCA突变的HER2 阴性乳腺癌患者,他们以 1:1 的比例随机接受每天两次 300 mg 口服奥拉帕利(n = 921)为期 1 年或安慰剂治疗(n = 915)。此外,患者需要接受早期(II-III 期)乳腺癌的治疗,并完成手术和化疗,无论是否接受放疗。纳入标准还要求患者具有疾病高复发风险,而那些先前接受过 PARP 抑制剂治疗的人没有资格参加。
该研究的主要终点是无侵袭性疾病生存(iDFS),而次要终点包括远处无病生存 (DDFS)、总生存 (OS)、健康相关的生活质量和安全性。

结果表明:在中位随访 2.5 年后,接受奥拉帕利治疗的患者减少了 42% iDFS(风险比 [HR]=0.58;99.5% CI=0.41-0.82;P <.0001 )。此外,研究人员们还注意到奥拉帕利和安慰剂之间的 3 年 iDFS 率差异为 8.8%(95% CI=4.5%-13.0%;分层HR=0.58;99.5% CI=0.41-0.82;P <.0001)。DDFS 降低了43%(HR=0.57;99.5% CI= 0.39-0.83;P <.0001),奥拉帕利和安慰剂的 3年DDFS率差异为 7.1%(95% CI=3.0%-11.1%;分层HR=0.57;99.5%=0.39-0.83;P <.0001)。

在中位随访 2.5 年时,接受奥拉帕尼与安慰剂的患者报告的死亡人数较少,两个研究队列之间的 OS 没有显著差异(HR=0.68;99% CI=0.44-1.05;P = .024);此外,两组之间的 3 年 OS 率差异为 3.7%(95% CI=0.3%-7.1%)。
就安全性而言,奥拉帕利组报告的不良反应 (AE) 与先前报告的结果一致。此外,奥拉帕利没有增加严重的 AE,包括住院或其他癌症(如白血病)的发生。在接受奥拉帕尼治疗的患者中≥3级AE包括贫血 (9%)、中性粒细胞减少 (5%)、白细胞减少 (3%) 和疲劳 (2%)。奥拉帕利组最常见的AE包括恶心 (57%)、疲劳 (40%)、贫血 (23%)、呕吐 (23%) 和头痛 (20%);而安慰剂组最常见的AE是疲劳 (27%)、恶心 (23%)、头痛 (17%)、腹泻 (14%) 和关节痛 (12%)。
二代 TROP2 ADC闪耀SABCS国际舞台,
Trodelvy耐药者有效!
Datopotamab deruxtecan为第一三共采用DXd ADC技术开发的新一代ADC药物。Datopotamab deruxtecan(Dato-DXd;DS-1062)通过一种4肽链接子将靶向肿瘤细胞表面特定抗原的人源化单克隆抗体(抗TROP2单抗)与一种新型拓扑异构酶1抑制剂exatecan衍生物(DX-8951衍生物,DXd)链接在一起。1期TROPIONPanTumor01研究的初步结果表明,DS-1062对非小细胞肺癌(NSCLC)(Meric-Bernstam,ASCO2021)和三阴性乳腺癌(TNBC)(Bardia,ESMOBC2021)患者具有令人鼓舞的抗肿瘤活性和可管理的安全性。
在 TNBC 队列中,44 名患者中有 42 名每 3 周接受 6 mg/kg 的DS-1062静脉注射 (IV),另外 2 名患者每 3 周接受 8 mg/kg 的 ADC IV。试验的主要终点是安全性和耐受性;次要终点包括疗效、药代动力学和抗药物抗体。
在试验的 TNBC 队列中的所有患者(n = 44)中,盲法独立中央审查 (BICR) 的 ORR 为 34%,中位随访时间为 7.6 个月(范围,4-13)。这包括 14 名患者 (32%) 确认的完全缓解 (CR) 或部分缓解 (PR),以及 1 名患者 (2%) 的 CR/PR 待确认。17 名患者 (39%) 疾病稳定 (SD),2 名患者 (5%) 不可评估 (NE),8 名患者 (18%) 疾病进展 (PD)。总体而言,疾病控制率 (DCR) 为 77%。

此外,在 8.8 个月(范围,4-13)的中位随访中,TNBC 患者之前未接受基于 Topo I 抑制剂的 ADC 治疗(n = 27)的 ORR 为 52%,其中包括13 名患者 (48%) 具有确认的 CR 或 PRS,另外 1 名患者 (4%) 具有 CR/PR 待确认。9 名患者 (33%) 患有 SD,1 名患者 (4%) 患有 NE,4 名患者 (15%) 患有 PD。DCR 为 81%。

此外,DS-1062显示出可控的安全性,没有新的安全信号。最常见的治疗出现的不良事件 (TEAE) 包括恶心、口腔炎、呕吐和疲劳。然而,中性粒细胞减少症和腹泻都不常见。没有观察到判定为治疗相关间质性肺病的病例。
98% 的患者至少有 1 次 TEAE,其中 45% 被认为是 3 级或更高;23% 的人经历了 3 级或更高级别的治疗相关 TEAE。18% 的患者需要因 AE 减少剂量,14% 的患者因 AE 出现治疗中断。一名患者因 AE 停止治疗,没有报告致命的 AE。
K药再发力!
全面布局TNBC,改善早/晚期患者生存期
7月27日,FDA正式批准帕博利珠单抗(Keytruda)用于治疗高危、早期三阴性乳腺癌(TNBC)患者,联合化疗作为新辅助治疗,然后继续作为单药作为手术后的辅助治疗。
KEYNOTE-522(NCT03036488)是一项3期研究,采用术前新辅助K药/安慰剂+化疗,然后术后辅助K药/安慰剂治疗早期TNBC患者。初步分析显示,加化疗后无事件生存期(EFS)有统计学意义和临床意义。
既往未治疗、非转移性、集中证实的TNBC患者(T1cN1-2或T2-4N0-2期每AJCC)按2:1随机分至K药200mgQ3W或安慰剂,均给予4个周期紫杉醇+卡铂,然后给予4个周期阿霉素或表阿霉素+环磷酰胺(新辅助期);手术后,患者接受K药或安慰剂9个周期,或直到复发或不可接受的毒性(辅助期)。
主要 EFS 分析的结果显示,784 名接受K药联合化疗随后接受K药治疗的患者发生 EFS 事件的发生率为 15.7%,而安慰剂联合化疗随后接受安慰剂组的 390 名患者发生 EFS 事件的发生率为 23.8%(HR 0.63;95% CI , 0.48-0.82; P = .00031)。两个队列的中位随访时间为 39.1 个月(范围,30.0-48.0)。36 个月的 EFS 率分别为 84.5% 和 76.8%。K药治疗方案获得的益处与 PD-L1 的表达状态无关。


在3期试验KEYNOTE-355(NCT02819518)中,K药联合化疗治疗既往未治疗的局部复发性不可手术或转移性TNBC患者,其肿瘤表达PD-L1,联合阳性评分(CPS)≥10。在CPS≥1人群中,治疗组间差异无统计学意义。本次报道了不同PD-L1表达情况下的PFS和OS结果。
共有847例患者按2:1随机分至K药+化疗(白蛋白紫杉醇100mg/m2天1、8和15天;紫杉醇90mg/m2天1、8天15;或吉西他滨1000mg/m2+卡铂AUC2天1和8每21天)或安慰剂+化疗组最多35次,或直至进展/无法忍受的毒性。
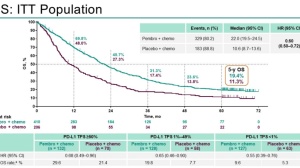
结果显示,在 CPS 为 10 或更高的患者中,K药和化疗联合治疗的中位 OS 为 23.0 个月,而单独化疗为 16.1 个月(HR,0.73;95% CI,0.55-0.95 ;P = .0093)。K药组的 18 个月 OS 率为 58.3%,而安慰剂/化疗组为 44.7%;此外,在 CPS 至少为 10 的患者中,该组合引起的中位 PFS 为 9.7 个月,而安慰剂组为 5.6 个月(HR,0.66;95% CI,0.50-0.88)。K药组的 12 个月 PFS 率为 39.1%,安慰剂组为 23.0%。且随着CPS表达的提高,OS获益逐渐明显。

K药目前已被 FDA 批准与化疗联合用于治疗局部复发性不可切除或转移性 TNBC 患者,这些患者的肿瘤表达 PD-L1(CPS ≥10)。而今年SABCS上发布的KEYNOTE-355 的最终结果支持这一决定,CPS≥10是个合适的阈值。
罗氏与FDA谈判后主动撤回T药TNBC适应症
8月30日,罗氏自主撤回了阿替利珠单抗用于联合白蛋白结合型紫杉醇化疗作为治疗肿瘤表达 PD-L1 阳性的三阴性乳腺癌 (TNBC) 患者的适应症。
大家可以关注公众号”三阴姐妹互助圈“下载三阴乳腺癌的治疗资料包和添加病友群,请扫下面的二维码。

该适应症的撤回是基于IMpassion031的研究结果。结果表明,58% (95% CI, 50%-65%) 的患者在接受T药治疗方案后出现病理完全缓解 (pCR),而单独接受化疗的患者为 41% (95% CI, 34%-49%) (95% CI,6%-27%;单方面P = .0044)。在具有 PD-L1 阳性的患者亚组中,研究组和对照组的PCR率分别为 69%(95% CI,57%-79%)和 49%(95% CI,38%-61%)。安全性数据与既往研究结果一致,未发现新的安全性信号。
“此次撤回是基于人用医药产品委员会认为提供的 IMpassion031 研究数据不允许委员会得出在拟议适应症中使用 [atezolizumab] 的积极利益风险平衡的结论。” Roche Registration GmbH 在给 EMA 的信中表示。
罗氏表示今后T药还会继续涉猎于三阴乳腺癌,期待它的喜报。
云顶新耀ADC获FDA批准上市,
CDE优先审评
云顶新耀和吉利德科学于近日联合公布,戈沙妥珠单抗(美国商品名:Trodelvy) 治疗转移性三阴性乳腺癌(TNBC)的IIb期临床试验(EVER-132-001)达到其总体缓解率(ORR)的主要终点。它是FDA批准的第一个用于复发性或难治性TNBC的抗体偶联药物,也是批准的第一个靶向TROP2的抗体偶联药物。目前国内尚无同靶点药物获批上市。
EVER-132-001是一项单臂、多中心的Ⅱb期注册研究,在中国招募的80例患者中评估戈沙妥珠单抗用于治疗既往接受过至少两种系统治疗(其中至少一种为针对转移性疾病的治疗)的不可切除的局部晚期或转移性三阴性乳腺癌成人患者的疗效。参与者在 21 天治疗周期的第 1 天和第 8 天以 10 mg/kg 的剂量静脉注射戈沙妥珠单抗。治疗一直持续到疾病进展、出现不可接受的毒性或撤回同意。
研究结果显示经独立审评委员会评估的总体缓解率(ORR)为38.8%(置信区间CI:95%)。此外,事实证明,该药剂的毒性特征与之前检查其使用的其他临床试验中报告的毒性特征相当。没有观察到新的安全信号。
“这些一线结果证实,sacituzumab govitecan 有可能帮助改变中国 mTNBC 患者的治疗前景,” Everest Medicines 肿瘤学首席医疗官杨石在一份新闻稿中表示;“这些数据,连同在全球 ASCENT 研究中看到的好处,支持其作为目前选择极其有限的患者的新型治疗方法的潜力。”
2020 年 5 月,中国国家药品监督管理局药品审评中心对戈沙妥珠单抗的生物制剂许可申请给予优先审评,适用于既往接受过 2 种或以上全身治疗的局部晚期或 mTNBC 成人患者,其中至少 1 种用于转移性治疗。

“Sacituzumab govitecan 对患者来说是一个重要的进步,因为它是全球首个获批用于 mTNBC 的 ADC,而 EVER-132-001 试验是实现我们对中国 mTNBC 患者承诺的重要的第一步,有意义地改善他们的生活,” Everest Medicines 首席执行官、医学博士、博士 Kerry Blanchard 在过去的新闻稿中表示。
在 2021 年 4 月,FDA 完全批准 sacituzumab govitecan用于先前接受过 2 种或多种全身治疗的不可切除局部晚期或 mTNBC 患者,其中至少 1 种用于可测量的疾病。
该获批是基于ASCENT是一项全球性随机3期试验,旨在对468例复发性或难治性转移性三阴性乳腺癌患者评价SG与单药化疗 (艾立布林、长春瑞滨、卡培他滨或吉西他滨,由医师选择) 相比的疗效和安全性。主要终点为未脑转移患者的无进展生存(由盲法独立集中复核确定),次要终点包括总生存、客观缓解率、安全性。结果发现,SG与化疗相比:
中位无进展生存:5.6月vs1.7月(95%置信区间:4.3~6.3、1.5~2.6)
进展或死亡风险:减少59%(风险比:0.41,95%置信区间:0.32~0.52,P<0.001)
中位总生存时间:12.1月vs6.7个月(95%置信区间:10.7~14.0、5.8~7.7)
总死亡风险:减少52%(风险比:0.48,95%置信区间:0.38~0.59,P<0.001)
客观缓解率:35%vs5%

3级或以上治疗相关不良事件包括:中性粒细胞减少(SG vs 化疗组:51% vs 33%)、白细胞减少(10% vs 5%)、腹泻(10% vs<1%)、贫血(8% vs 5%)和发热性中性粒细胞减少(6% vs 2%)。6名患者因不良事件死亡事件,但未发生与SG治疗有关的患者死亡。
新型单抗靶向CCR5获FDA认定,
OS超过12个月,
打破三阴乳腺癌无靶点定律!
近日,根据CytoDyn公司发布的公告显示,该司已经为其研发的CCR5拮抗剂Leronlimab(PRO140)向FDA提交了突破性疗法的申请,作为转移性三阴性乳腺癌的一种潜在治疗方案。更值得一提的是,FDA已授予CCR5拮抗剂leronlimab(PRO140)的快速通道认证,用于与卡铂联合用于治疗CCR5阳性的转移性三阴性乳腺癌患者。
该申请的提交基于一项28例转移性三阴性乳腺癌患者的试验结果。这些患者在接受Leronlimab的治疗之前,已经接受过至少2种方案的治疗并失败。
这些患者来自3项临床试验(NCT04313075,NCT03838367以及一项篮子试验),均为Ⅳ期转移性三阴性乳腺癌。在接受治疗前,61%的患者存在内脏转移,75%的患者存在脑转移,11%的患者存在骨转移。
试验结果显示,接受Leronlimab治疗的患者,中位总生存期(mOS)超过了12个月!显著优于SOC化疗组(6.6个月)或SacituzumabGovitecan(SG)组(11.8个月)。
在无进展生存(mPFS)方面,接受更高剂量(≥525mg)Leronlimab联合化疗的患者的mPFS为6.2个月(95%CI,2.6–7.5个月),与SOC化疗(2.3个月)和SG(4.8个月)相比,中位无进展生存期也得到显著延长。
响应率方面Leronlimab的表现同样稳定,疾病控制率达到了92%。
美国正式启动
首个预防型乳腺癌疫苗I期临床试验
近日,美国克利夫兰诊所的研究人员正式启动了一项最新的疫苗研究,目的是最终能为健康女性预防三阴性乳腺癌这种极具侵袭性和致命性的癌症。如果能够成功,将造福所有女性,从此终结三阴乳癌的噩梦!

该 I 期试验旨在确定早期三阴性乳腺癌患者的疫苗最大耐受剂量,并表征和优化身体的免疫反应。美国食品和药物管理局最近批准了该疫苗的一项研究性新药申请,这允许克利夫兰诊所和合作伙伴 Anixa Biosciences, Inc.(ANIX:纳斯达克)开始这项研究。
这项新研究是基于此前Tuohy领导的一项临床前研究,该研究表明,激活靶向α-乳清蛋白的免疫系统在预防小鼠乳腺肿瘤方面是安全有效的。研究还发现,单次疫苗接种就可以防止小鼠模型发生乳腺肿瘤,同时还能抑制已经存在的乳腺肿瘤生长。该研究最初发表在《Nature Medicine》上,研究部分资金来自过去12年里超过2万人的慈善捐赠。
“这种疫苗方法代表了一种控制乳腺癌的潜在新方法,”该疫苗的主要发明者和克利夫兰诊所勒纳研究所的工作人员免疫学家Vincent Tuohy 博士说;“这项研究的长期目标是确定这种疫苗是否可以在乳腺癌发生之前预防它,尤其是在高危女性中占主导地位的更具侵袭性的这种疾病。”
该研究性疫苗针对的是一种乳腺特异性泌乳蛋白 α-乳清蛋白,该蛋白在正常、老化的组织中不再存在于泌乳后,但存在于大多数三阴性乳腺癌中。激活针对这种“退休”蛋白质的免疫系统可提供针对表达α-乳清蛋白的新兴乳腺肿瘤的先发制人的免疫保护。该疫苗还含有一种佐剂,可激活先天免疫反应,使免疫系统对新出现的肿瘤产生反应,以防止它们生长。
研究人员预计,随后的试验将涉及具有患乳腺癌高风险的健康、无癌症女性,这些女性已决定自愿接受双侧乳房切除术以降低风险。通常,这些女性携带BRCA1或BRCA2基因突变,因此有患三阴性乳腺癌的风险,或者有患任何形式乳腺癌的高家族风险。
“这种疫苗策略有可能应用于其他肿瘤类型,”Tuohy 博士补充道。“我们的转化研究计划侧重于开发疫苗,以预防我们因年龄增长而面临的疾病,如乳腺癌、卵巢癌和子宫内膜癌。如果成功,这些疫苗有可能改变我们控制成人发病的癌症的方式,并以类似于儿童疫苗接种计划产生的影响的方式延长预期寿命。”
FDA授予trilaciclib乳腺癌快速通道,
为化疗保驾护航!
7月20日,FDA 已授予 trilaciclib (Cosela)与化疗联合用于局部晚期或转移性三阴性乳腺癌 (TNBC) 患者的快速通道指定。此前,Trilaciclib于2021年2月12日已获得FDA批准上市,用于小细胞肺癌化疗引起的骨髓抑制,并纳入2021年V3版NCCN小细胞肺癌指南推荐,成功成为首个被推荐作为化疗前骨髓预防的标准疗法,同时也是全球首款骨髓保护剂!
在一项 2 期试验 (NCT02978716) 中纳入了美国26个地区的活检确认的局部复发或转移性三阴性乳腺癌成人患者,之前至多接受过两线化疗。
符合条件患者随机分配为三组接受治疗:
1组接受吉西他滨+卡铂;
2组接受吉西他滨+卡铂+trilaciclib(240mg/m2;第1、8天);
3组接受接受吉西他滨+卡铂(第2、9天)+trilaciclib(第1、2、8、9天),一个周期21天。
主要研究终点为联合治疗的安全性和耐受性。结果显示,联合组化疗暴露的时间更长,而且与普通化疗组相比,联合组发生不良事件的发生率相同或更低。虽然尚未达到骨髓保护的主要终点,但 trilaciclib 确实显著提高了总体生存率 (OS),且无论 PD-L1 状态如何。在疗效可评估患者中,三组的ORR分别为33%、50%和37%。与普通化疗组相比,两个试验组的PFS和OS均显著延长,PFS分别为:5.7月:9.41月:7.3月;OS分别为12.6月:20.1月:17.8月。亚组分析中观察到的PFS和OS的获益于总人群一致。

该药物目前正在作为多中心、双盲、安慰剂对照的 3 期 PRESERVE 2 试验 (NCT04799249) 的一部分进行评估。旨再一次确认在 2 期试验中观察到的 OS 获益,并进一步检查该方法在检查点后抑制剂患者群体中的疗效。该试验于 2021 年 4 月启动,预计数据将于 2023 年下半年公布。
本文仅供医学药学专业人士阅读


.jpg-pd13)


















 X
X