
 双特异性抗体(红色箭头)引导T细胞攻击肿瘤细胞
双特异性抗体(红色箭头)引导T细胞攻击肿瘤细胞
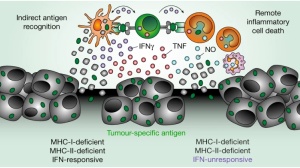
 靶向特定TCR的双特异性抗体迅速清除小鼠体内肿瘤(中),而无关抗体(左)和敲除肿瘤TCR(右)都使肿瘤未被清除
靶向特定TCR的双特异性抗体迅速清除小鼠体内肿瘤(中),而无关抗体(左)和敲除肿瘤TCR(右)都使肿瘤未被清除 双特异性抗体使携带TP53突变的肿瘤消退
双特异性抗体使携带TP53突变的肿瘤消退 双特异性抗体延缓了白血病细胞HL-60的生长
双特异性抗体延缓了白血病细胞HL-60的生长点击查看全文
本文仅供医学药学专业人士阅读
双特异性抗体(红色箭头)引导T细胞攻击肿瘤细胞靶向特定TCR的双特异性抗体迅速清除小鼠体内肿瘤(中),而无关抗体(左)和敲除肿瘤TCR(右)都使肿瘤未被清除双特异性抗体使携带TP53突变的肿瘤消退双特异性抗体延缓了白血病细胞HL-60的生长
本文仅供医学药学专业人士阅读

 top1
top1 top2
top2 top3
top3 top4
top4 top5
top5 top6
top6

癌友圈公众号

咚咚肿瘤科app

咚咚健康小程序








 X
X